Chapter 12 Bach mouse mammary gland (10X Genomics)
12.1 Introduction
This performs an analysis of the Bach et al. (2017) 10X Genomics dataset, from which we will consider a single sample of epithelial cells from the mouse mammary gland during gestation.
12.3 Quality control
is.mito <- rowData(sce.mam)$SEQNAME == "MT"
stats <- perCellQCMetrics(sce.mam, subsets=list(Mito=which(is.mito)))
qc <- quickPerCellQC(stats, percent_subsets="subsets_Mito_percent")
sce.mam <- sce.mam[,!qc$discard]colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- qc$discard
gridExtra::grid.arrange(
plotColData(unfiltered, y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
ncol=2
)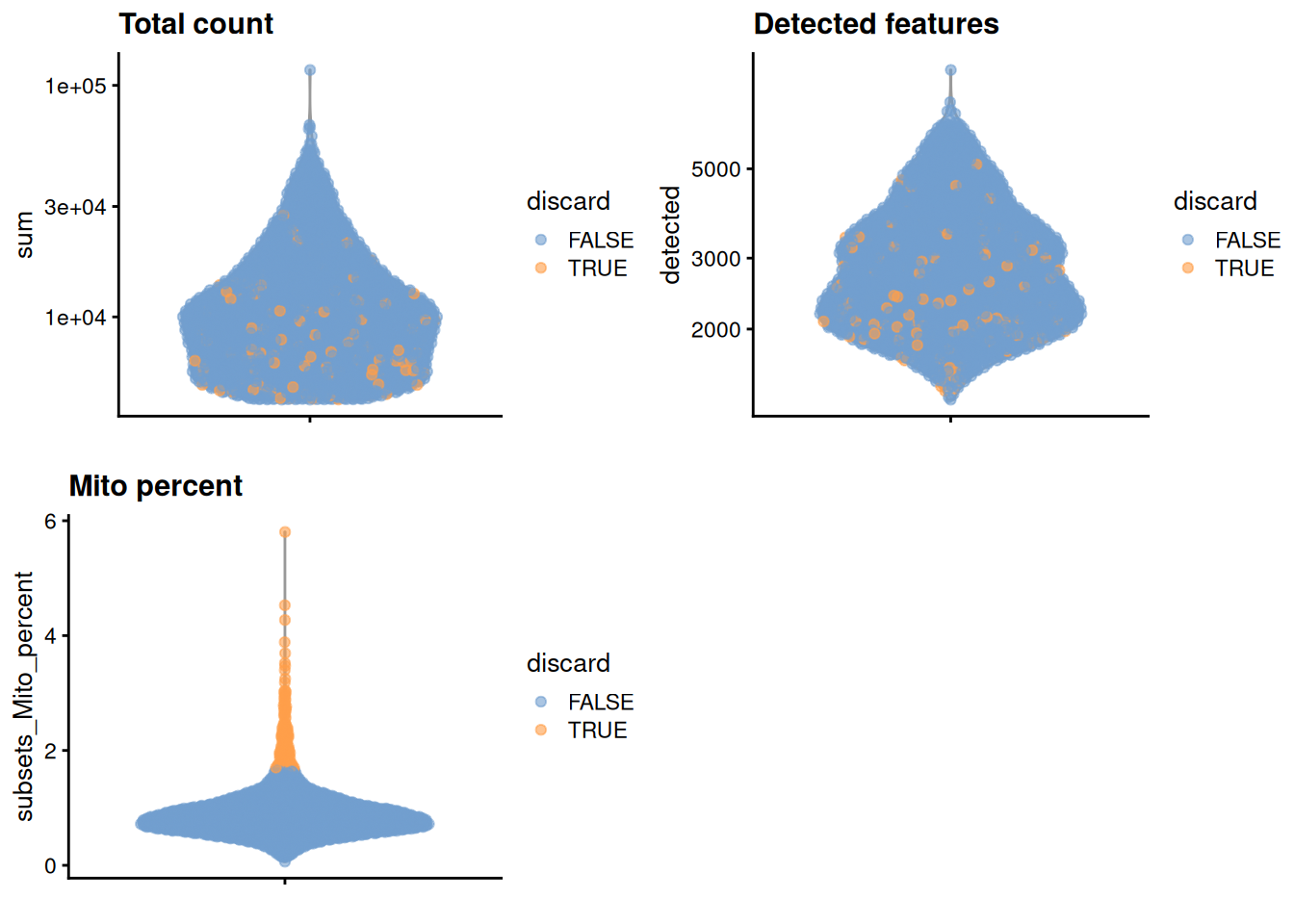
Figure 12.1: Distribution of each QC metric across cells in the Bach mammary gland dataset. Each point represents a cell and is colored according to whether that cell was discarded.
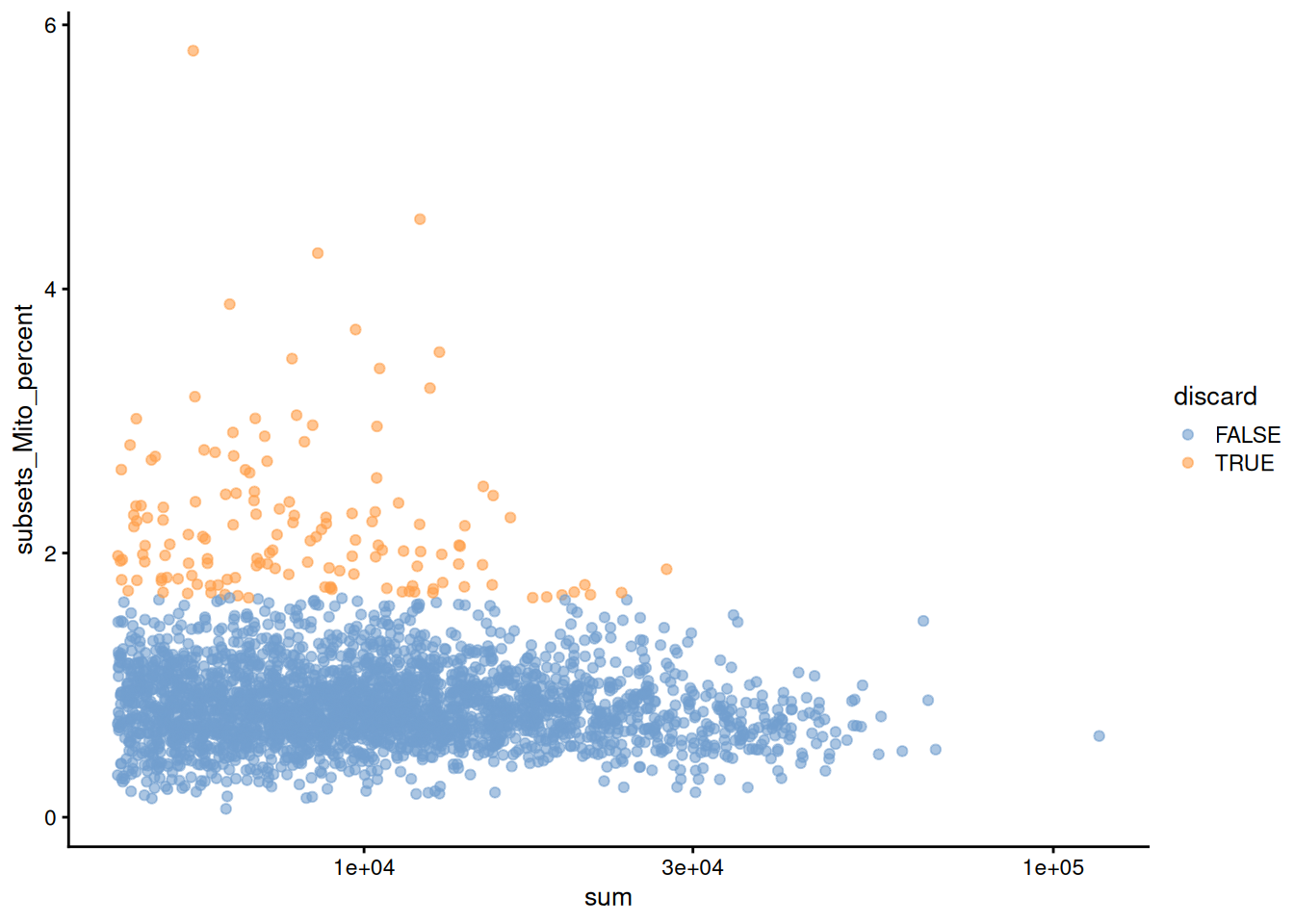
Figure 12.2: Percentage of mitochondrial reads in each cell in the Bach mammary gland dataset compared to its total count. Each point represents a cell and is colored according to whether that cell was discarded.
## low_lib_size low_n_features high_subsets_Mito_percent
## 0 0 143
## discard
## 14312.4 Normalization
library(scran)
set.seed(101000110)
clusters <- quickCluster(sce.mam)
sce.mam <- computeSumFactors(sce.mam, clusters=clusters)
sce.mam <- logNormCounts(sce.mam)## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.264 0.520 0.752 1.000 1.207 10.790plot(librarySizeFactors(sce.mam), sizeFactors(sce.mam), pch=16,
xlab="Library size factors", ylab="Deconvolution factors", log="xy")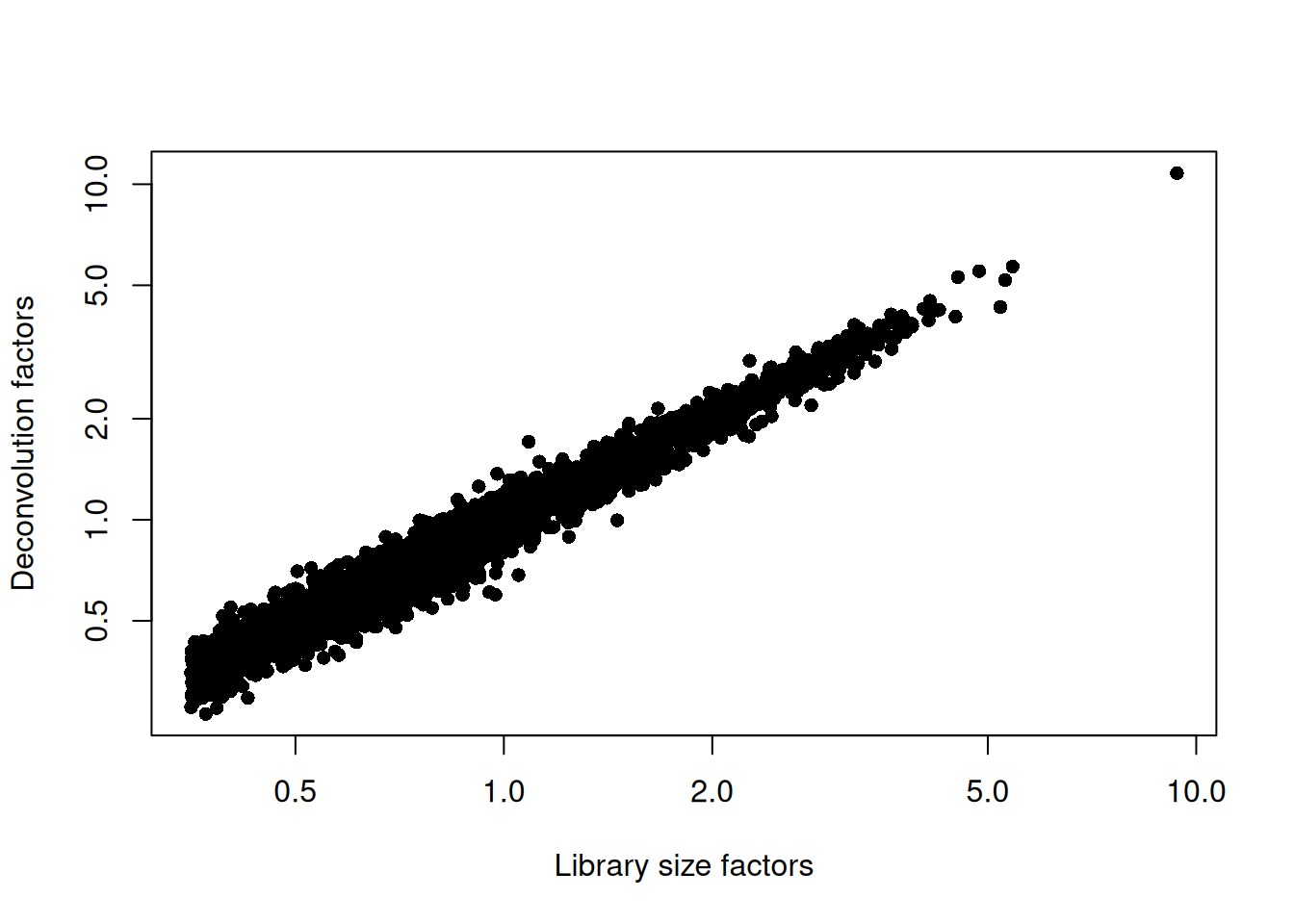
Figure 12.3: Relationship between the library size factors and the deconvolution size factors in the Bach mammary gland dataset.
12.5 Variance modelling
We use a Poisson-based technical trend to capture more genuine biological variation in the biological component.
set.seed(00010101)
dec.mam <- modelGeneVarByPoisson(sce.mam)
top.mam <- getTopHVGs(dec.mam, prop=0.1)plot(dec.mam$mean, dec.mam$total, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(dec.mam)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)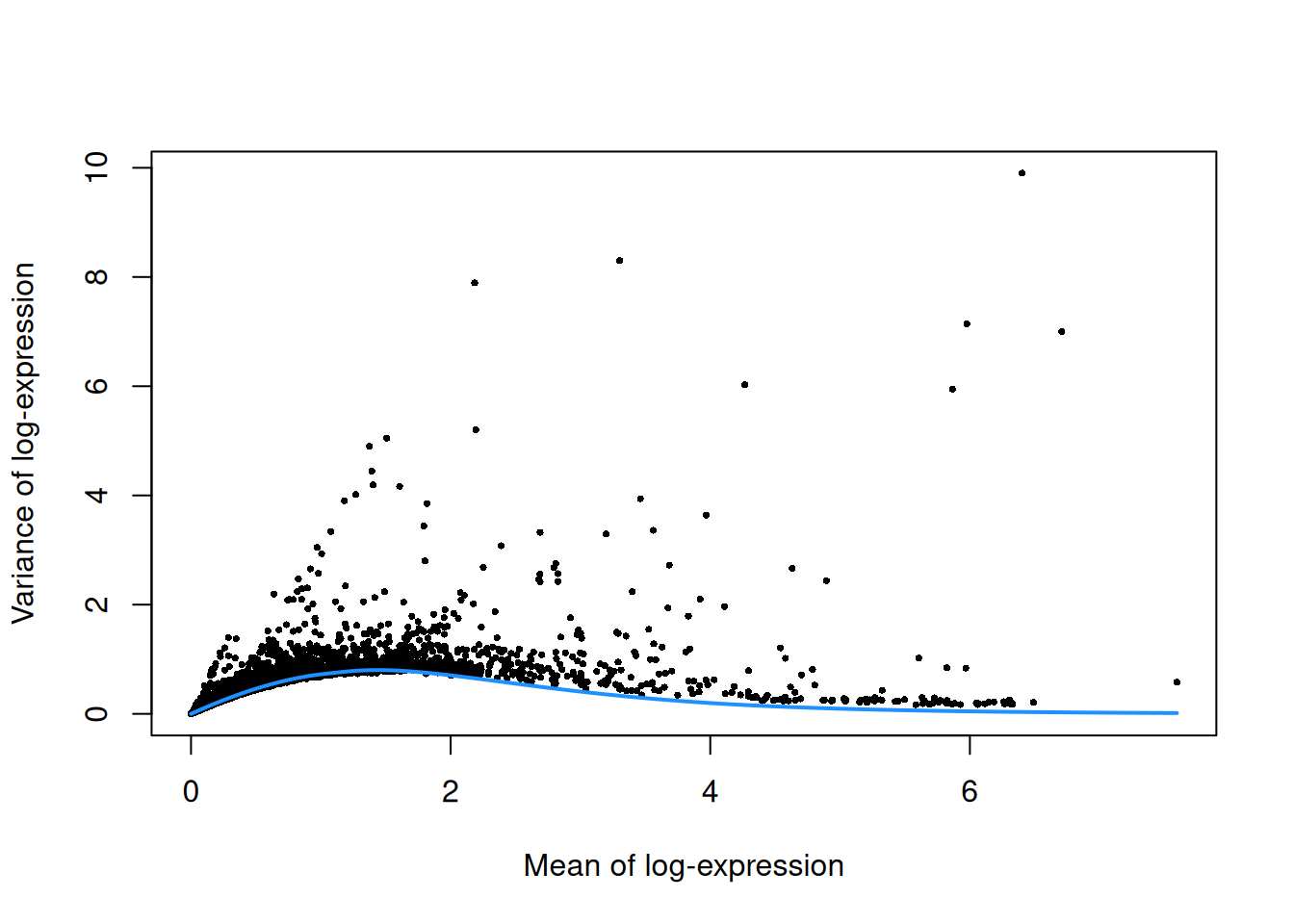
Figure 12.4: Per-gene variance as a function of the mean for the log-expression values in the Bach mammary gland dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to simulated Poisson counts.
12.6 Dimensionality reduction
library(BiocSingular)
set.seed(101010011)
sce.mam <- denoisePCA(sce.mam, technical=dec.mam, subset.row=top.mam)
sce.mam <- runTSNE(sce.mam, dimred="PCA")## [1] 1512.7 Clustering
We use a higher k to obtain coarser clusters (for use in doubletCluster() later).
snn.gr <- buildSNNGraph(sce.mam, use.dimred="PCA", k=25)
colLabels(sce.mam) <- factor(igraph::cluster_walktrap(snn.gr)$membership)##
## 1 2 3 4 5 6 7 8 9 10
## 550 847 639 477 54 88 39 22 32 24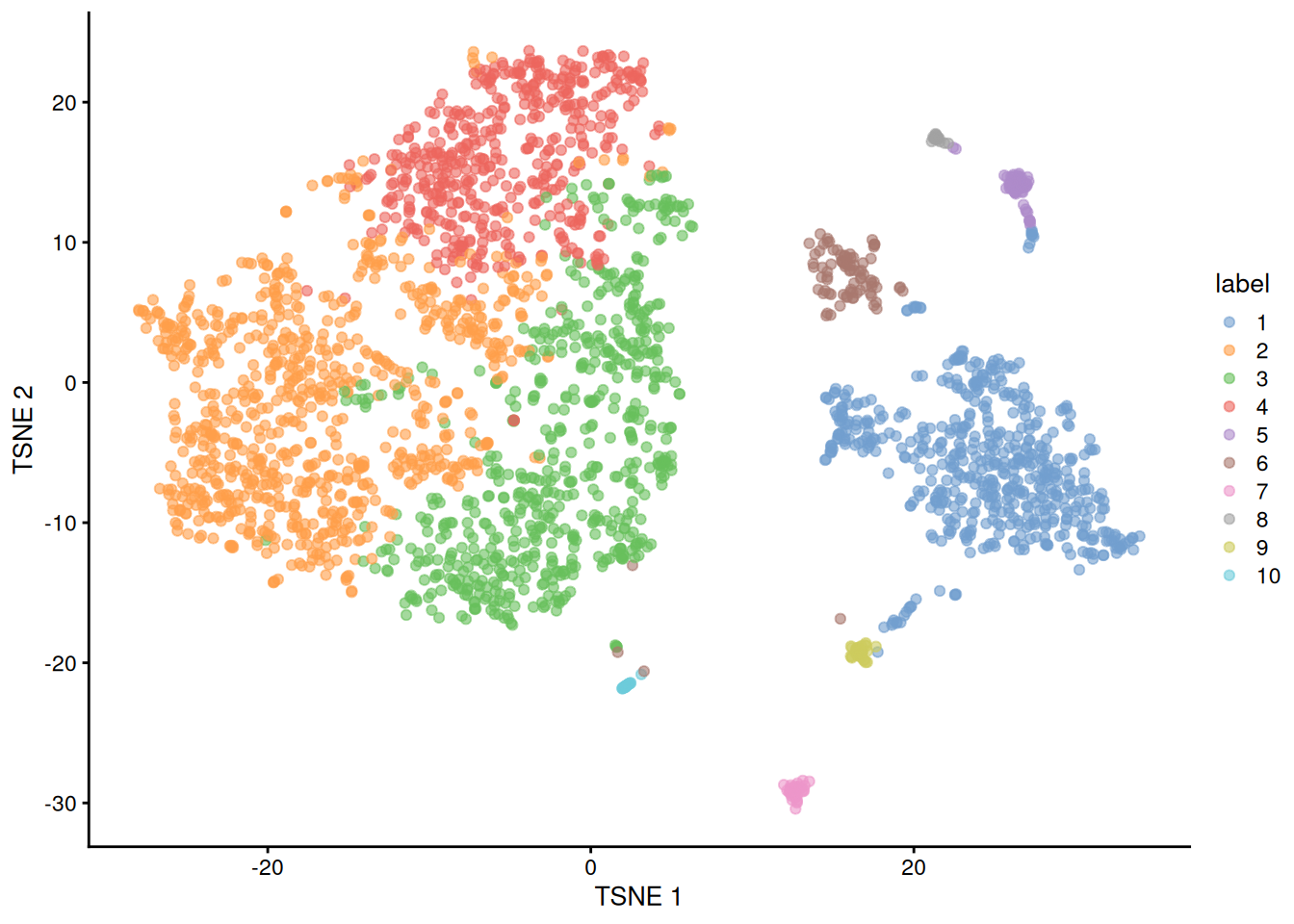
Figure 12.5: Obligatory \(t\)-SNE plot of the Bach mammary gland dataset, where each point represents a cell and is colored according to the assigned cluster.
Session Info
R version 4.4.1 (2024-06-14)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 24.04.1 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.20-bioc/R/lib/libRblas.so
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.12.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: America/New_York
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] BiocSingular_1.22.0 scran_1.34.0
[3] AnnotationHub_3.14.0 BiocFileCache_2.14.0
[5] dbplyr_2.5.0 scater_1.34.0
[7] ggplot2_3.5.1 scuttle_1.16.0
[9] ensembldb_2.30.0 AnnotationFilter_1.30.0
[11] GenomicFeatures_1.58.0 AnnotationDbi_1.68.0
[13] scRNAseq_2.19.1 SingleCellExperiment_1.28.0
[15] SummarizedExperiment_1.36.0 Biobase_2.66.0
[17] GenomicRanges_1.58.0 GenomeInfoDb_1.42.0
[19] IRanges_2.40.0 S4Vectors_0.44.0
[21] BiocGenerics_0.52.0 MatrixGenerics_1.18.0
[23] matrixStats_1.4.1 BiocStyle_2.34.0
[25] rebook_1.16.0
loaded via a namespace (and not attached):
[1] jsonlite_1.8.9 CodeDepends_0.6.6 magrittr_2.0.3
[4] ggbeeswarm_0.7.2 gypsum_1.2.0 farver_2.1.2
[7] rmarkdown_2.28 BiocIO_1.16.0 zlibbioc_1.52.0
[10] vctrs_0.6.5 memoise_2.0.1 Rsamtools_2.22.0
[13] RCurl_1.98-1.16 htmltools_0.5.8.1 S4Arrays_1.6.0
[16] curl_5.2.3 BiocNeighbors_2.0.0 Rhdf5lib_1.28.0
[19] SparseArray_1.6.0 rhdf5_2.50.0 sass_0.4.9
[22] alabaster.base_1.6.0 bslib_0.8.0 alabaster.sce_1.6.0
[25] httr2_1.0.5 cachem_1.1.0 GenomicAlignments_1.42.0
[28] igraph_2.1.1 mime_0.12 lifecycle_1.0.4
[31] pkgconfig_2.0.3 rsvd_1.0.5 Matrix_1.7-1
[34] R6_2.5.1 fastmap_1.2.0 GenomeInfoDbData_1.2.13
[37] digest_0.6.37 colorspace_2.1-1 dqrng_0.4.1
[40] irlba_2.3.5.1 ExperimentHub_2.14.0 RSQLite_2.3.7
[43] beachmat_2.22.0 labeling_0.4.3 filelock_1.0.3
[46] fansi_1.0.6 httr_1.4.7 abind_1.4-8
[49] compiler_4.4.1 bit64_4.5.2 withr_3.0.2
[52] BiocParallel_1.40.0 viridis_0.6.5 DBI_1.2.3
[55] highr_0.11 HDF5Array_1.34.0 alabaster.ranges_1.6.0
[58] alabaster.schemas_1.6.0 rappdirs_0.3.3 DelayedArray_0.32.0
[61] bluster_1.16.0 rjson_0.2.23 tools_4.4.1
[64] vipor_0.4.7 beeswarm_0.4.0 glue_1.8.0
[67] restfulr_0.0.15 rhdf5filters_1.18.0 grid_4.4.1
[70] Rtsne_0.17 cluster_2.1.6 generics_0.1.3
[73] gtable_0.3.6 metapod_1.14.0 ScaledMatrix_1.14.0
[76] utf8_1.2.4 XVector_0.46.0 ggrepel_0.9.6
[79] BiocVersion_3.20.0 pillar_1.9.0 limma_3.62.0
[82] dplyr_1.1.4 lattice_0.22-6 rtracklayer_1.66.0
[85] bit_4.5.0 tidyselect_1.2.1 locfit_1.5-9.10
[88] Biostrings_2.74.0 knitr_1.48 gridExtra_2.3
[91] bookdown_0.41 ProtGenerics_1.38.0 edgeR_4.4.0
[94] xfun_0.48 statmod_1.5.0 UCSC.utils_1.2.0
[97] lazyeval_0.2.2 yaml_2.3.10 evaluate_1.0.1
[100] codetools_0.2-20 tibble_3.2.1 alabaster.matrix_1.6.0
[103] BiocManager_1.30.25 graph_1.84.0 cli_3.6.3
[106] munsell_0.5.1 jquerylib_0.1.4 Rcpp_1.0.13
[109] dir.expiry_1.14.0 png_0.1-8 XML_3.99-0.17
[112] parallel_4.4.1 blob_1.2.4 bitops_1.0-9
[115] viridisLite_0.4.2 alabaster.se_1.6.0 scales_1.3.0
[118] purrr_1.0.2 crayon_1.5.3 rlang_1.1.4
[121] cowplot_1.1.3 KEGGREST_1.46.0 References
Bach, K., S. Pensa, M. Grzelak, J. Hadfield, D. J. Adams, J. C. Marioni, and W. T. Khaled. 2017. “Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing.” Nat Commun 8 (1): 2128.