Chapter 3 Feature selection
3.1 Motivation
We often use scRNA-seq data in exploratory analyses to characterize heterogeneity across cells. Procedures like clustering and dimensionality reduction compare cells based on their gene expression profiles, which involves aggregating per-gene differences into a single (dis)similarity metric between a pair of cells. The choice of genes to use in this calculation has a major impact on the behavior of the metric and the performance of downstream methods. We want to select genes that contain useful information about the biology of the system while removing genes that contain random noise. This aims to preserve interesting biological structure without the variance that obscures that structure, and to reduce the size of the data to improve computational efficiency of later steps.
The simplest approach to feature selection is to select the most variable genes based on their expression across the population. This assumes that genuine biological differences will manifest as increased variation in the affected genes, compared to other genes that are only affected by technical noise or a baseline level of “uninteresting” biological variation (e.g., from transcriptional bursting). Several methods are available to quantify the variation per gene and to select an appropriate set of highly variable genes (HVGs). We will discuss these below using the 10X PBMC dataset for demonstration:
#--- loading ---#
library(DropletTestFiles)
raw.path <- getTestFile("tenx-2.1.0-pbmc4k/1.0.0/raw.tar.gz")
out.path <- file.path(tempdir(), "pbmc4k")
untar(raw.path, exdir=out.path)
library(DropletUtils)
fname <- file.path(out.path, "raw_gene_bc_matrices/GRCh38")
sce.pbmc <- read10xCounts(fname, col.names=TRUE)
#--- gene-annotation ---#
library(scater)
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol)
library(EnsDb.Hsapiens.v86)
location <- mapIds(EnsDb.Hsapiens.v86, keys=rowData(sce.pbmc)$ID,
column="SEQNAME", keytype="GENEID")
#--- cell-detection ---#
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
sce.pbmc <- sce.pbmc[,which(e.out$FDR <= 0.001)]
#--- quality-control ---#
stats <- perCellQCMetrics(sce.pbmc, subsets=list(Mito=which(location=="MT")))
high.mito <- isOutlier(stats$subsets_Mito_percent, type="higher")
sce.pbmc <- sce.pbmc[,!high.mito]
#--- normalization ---#
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.pbmc)
sce.pbmc <- computeSumFactors(sce.pbmc, cluster=clusters)
sce.pbmc <- logNormCounts(sce.pbmc)## class: SingleCellExperiment
## dim: 33694 4080
## metadata(1): Samples
## assays(2): counts logcounts
## rownames(33694): RP11-34P13.3 FAM138A ... AC213203.1 FAM231B
## rowData names(2): ID Symbol
## colnames(4080): AAACCTGAGAAGGCCT-1 AAACCTGAGACAGACC-1 ...
## TTTGTCAGTTAAGACA-1 TTTGTCATCCCAAGAT-1
## colData names(3): Sample Barcode sizeFactor
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):As well as the 416B dataset:
#--- loading ---#
library(scRNAseq)
sce.416b <- LunSpikeInData(which="416b")
sce.416b$block <- factor(sce.416b$block)
#--- gene-annotation ---#
library(AnnotationHub)
ens.mm.v97 <- AnnotationHub()[["AH73905"]]
rowData(sce.416b)$ENSEMBL <- rownames(sce.416b)
rowData(sce.416b)$SYMBOL <- mapIds(ens.mm.v97, keys=rownames(sce.416b),
keytype="GENEID", column="SYMBOL")
rowData(sce.416b)$SEQNAME <- mapIds(ens.mm.v97, keys=rownames(sce.416b),
keytype="GENEID", column="SEQNAME")
library(scater)
rownames(sce.416b) <- uniquifyFeatureNames(rowData(sce.416b)$ENSEMBL,
rowData(sce.416b)$SYMBOL)
#--- quality-control ---#
mito <- which(rowData(sce.416b)$SEQNAME=="MT")
stats <- perCellQCMetrics(sce.416b, subsets=list(Mt=mito))
qc <- quickPerCellQC(stats, percent_subsets=c("subsets_Mt_percent",
"altexps_ERCC_percent"), batch=sce.416b$block)
sce.416b <- sce.416b[,!qc$discard]
#--- normalization ---#
library(scran)
sce.416b <- computeSumFactors(sce.416b)
sce.416b <- logNormCounts(sce.416b)## class: SingleCellExperiment
## dim: 46604 185
## metadata(0):
## assays(2): counts logcounts
## rownames(46604): 4933401J01Rik Gm26206 ... CAAA01147332.1
## CBFB-MYH11-mcherry
## rowData names(4): Length ENSEMBL SYMBOL SEQNAME
## colnames(185): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
## SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
## SLX-11312.N712_S507.H5H5YBBXX.s_8.r_1
## SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
## colData names(9): cell line cell type ... block sizeFactor
## reducedDimNames(0):
## mainExpName: endogenous
## altExpNames(2): ERCC SIRV3.2 Quantifying per-gene variation
The simplest approach to quantifying per-gene variation is to compute the variance of the log-normalized expression values (i.e., “log-counts” ) for each gene across all cells (A. T. L. Lun, McCarthy, and Marioni 2016). The advantage of this approach is that the feature selection is based on the same log-values that are used for later downstream steps. In particular, genes with the largest variances in log-values will contribute most to the Euclidean distances between cells during procedures like clustering and dimensionality reduction. By using log-values here, we ensure that our quantitative definition of heterogeneity is consistent throughout the entire analysis.
Calculation of the per-gene variance is simple but feature selection requires modelling of the mean-variance relationship.
The log-transformation is not a variance stabilizing transformation in most cases,
which means that the total variance of a gene is driven more by its abundance than its underlying biological heterogeneity.
To account for this effect, we use the modelGeneVar() function to fit a trend to the variance with respect to abundance across all genes (Figure 3.1).
library(scran)
dec.pbmc <- modelGeneVar(sce.pbmc)
# Visualizing the fit:
fit.pbmc <- metadata(dec.pbmc)
plot(fit.pbmc$mean, fit.pbmc$var, xlab="Mean of log-expression",
ylab="Variance of log-expression")
curve(fit.pbmc$trend(x), col="dodgerblue", add=TRUE, lwd=2)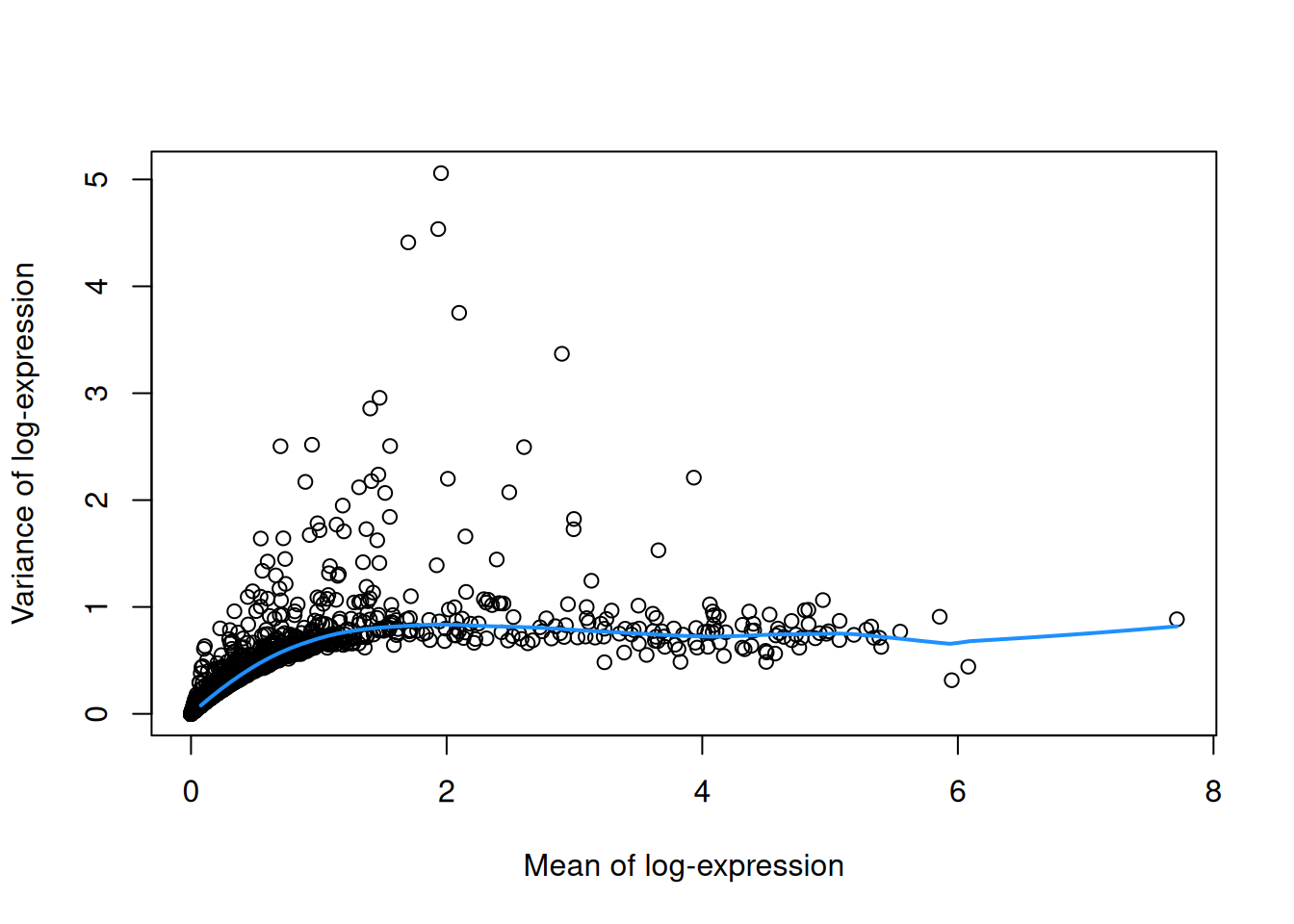
Figure 3.1: Variance in the PBMC data set as a function of the mean. Each point represents a gene while the blue line represents the trend fitted to all genes.
At any given abundance, we assume that the variation in expression for most genes is driven by uninteresting processes like sampling noise. Under this assumption, the fitted value of the trend at any given gene’s abundance represents an estimate of its uninteresting variation, which we call the technical component. We then define the biological component for each gene as the difference between its total variance and the technical component. This biological component represents the “interesting” variation for each gene and can be used as the metric for HVG selection.
# Ordering by most interesting genes for inspection.
dec.pbmc[order(dec.pbmc$bio, decreasing=TRUE),] ## DataFrame with 33694 rows and 6 columns
## mean total tech bio p.value FDR
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## LYZ 1.98587 5.13788 0.846825 4.29105 3.61154e-263 2.37062e-259
## S100A9 1.95667 4.60893 0.847197 3.76173 1.24604e-202 3.50530e-199
## S100A8 1.73216 4.52022 0.838745 3.68148 4.60805e-198 1.13427e-194
## HLA-DRA 2.10832 3.71606 0.842381 2.87367 1.46232e-120 1.37124e-117
## CD74 2.89589 3.35549 0.803978 2.55151 1.20427e-104 6.77555e-102
## ... ... ... ... ... ... ...
## HLA-B 4.46672 0.502751 0.747677 -0.244926 0.987426 1
## PTMA 3.80253 0.486855 0.742256 -0.255401 0.990662 1
## TMSB4X 6.06401 0.436253 0.695625 -0.259372 0.994593 1
## EIF1 3.21300 0.491514 0.780550 -0.289036 0.994315 1
## B2M 5.92342 0.317036 0.663622 -0.346586 0.999821 1(Careful readers will notice that some genes have negative biological components, which have no obvious interpretation and can be ignored in most applications. They are inevitable when fitting a trend to the per-gene variances as approximately half of the genes will lie below the trend.)
Strictly speaking, the interpretation of the fitted trend as the technical component assumes that the expression profiles of most genes are dominated by random technical noise. In practice, all expressed genes will exhibit some non-zero level of biological variability due to events like transcriptional bursting. Thus, it would be more appropriate to consider these estimates as technical noise plus “uninteresting” biological variation, under the assumption that most genes do not participate in the processes driving interesting heterogeneity across the population.
3.3 Quantifying technical noise
The assumption in Section 3.2 may be problematic in rare scenarios where many genes at a particular abundance are affected by a biological process. For example, strong upregulation of cell type-specific genes may result in an enrichment of HVGs at high abundances. This would inflate the fitted trend in that abundance interval and compromise the detection of the relevant genes. We can avoid this problem by fitting a mean-dependent trend to the variance of the spike-in transcripts (Figure 3.2), if they are available. The premise here is that spike-ins should not be affected by biological variation, so the fitted value of the spike-in trend should represent a better estimate of the technical component for each gene.
dec.spike.416b <- modelGeneVarWithSpikes(sce.416b, "ERCC")
dec.spike.416b[order(dec.spike.416b$bio, decreasing=TRUE),]## DataFrame with 46604 rows and 6 columns
## mean total tech bio p.value FDR
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## Lyz2 6.61097 13.8497 1.57131 12.2784 1.51773e-186 1.57032e-183
## Ccl9 6.67846 13.1869 1.50035 11.6866 2.25908e-185 2.23998e-182
## Top2a 5.81024 14.1787 2.54778 11.6309 3.83585e-65 1.14102e-62
## Cd200r3 4.83180 15.5613 4.22990 11.3314 9.50102e-24 6.11070e-22
## Ccnb2 5.97776 13.1393 2.30179 10.8375 3.72198e-69 1.21331e-66
## ... ... ... ... ... ... ...
## Rpl5-ps2 3.60625 0.612623 6.32854 -5.71591 0.999616 0.999726
## Gm11942 3.38768 0.798570 6.51471 -5.71614 0.999459 0.999726
## Gm12816 2.91276 0.838670 6.57355 -5.73488 0.999422 0.999726
## Gm13623 2.72844 0.708071 6.45440 -5.74633 0.999544 0.999726
## Rps12l1 3.15420 0.746615 6.59326 -5.84664 0.999522 0.999726plot(dec.spike.416b$mean, dec.spike.416b$total, xlab="Mean of log-expression",
ylab="Variance of log-expression")
fit.spike.416b <- metadata(dec.spike.416b)
points(fit.spike.416b$mean, fit.spike.416b$var, col="red", pch=16)
curve(fit.spike.416b$trend(x), col="dodgerblue", add=TRUE, lwd=2)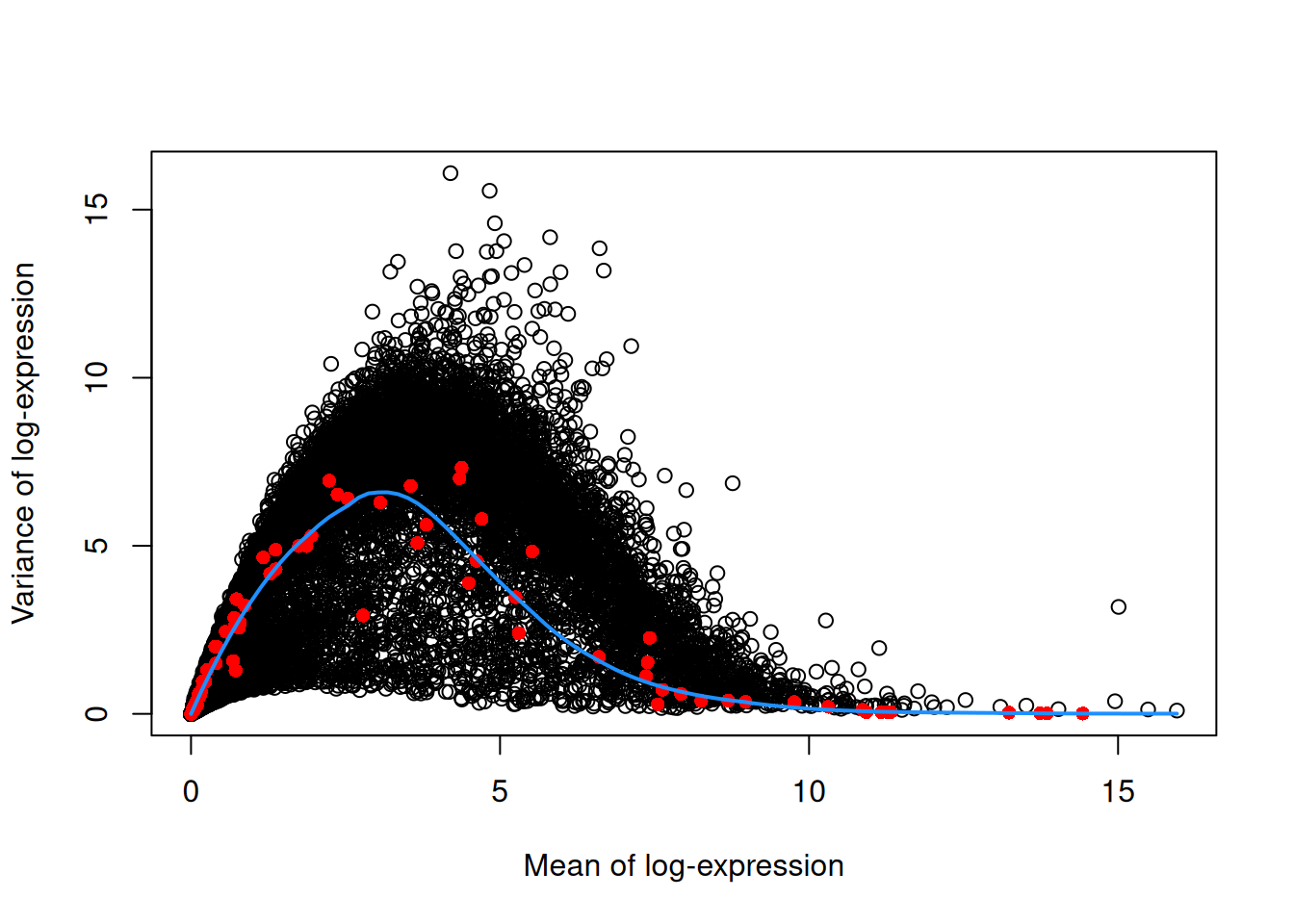
Figure 3.2: Variance in the 416B data set as a function of the mean. Each point represents a gene (black) or spike-in transcript (red) and the blue line represents the trend fitted to all spike-ins.
In the absence of spike-in data, one can attempt to create a trend by making some distributional assumptions about the noise.
For example, UMI counts typically exhibit near-Poisson variation if we only consider technical noise from library preparation and sequencing.
This can be used to construct a mean-variance trend in the log-counts (Figure 3.3) with the modelGeneVarByPoisson() function.
Note the increased residuals of the high-abundance genes, which can be interpreted as the amount of biological variation that was assumed to be “uninteresting” when fitting the gene-based trend in Figure 3.1.
set.seed(0010101)
dec.pois.pbmc <- modelGeneVarByPoisson(sce.pbmc)
dec.pois.pbmc <- dec.pois.pbmc[order(dec.pois.pbmc$bio, decreasing=TRUE),]
head(dec.pois.pbmc)## DataFrame with 6 rows and 6 columns
## mean total tech bio p.value FDR
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## LYZ 1.98587 5.13788 0.628429 4.50945 0 0
## S100A9 1.95667 4.60893 0.633955 3.97497 0 0
## S100A8 1.73216 4.52022 0.671689 3.84854 0 0
## HLA-DRA 2.10832 3.71606 0.604255 3.11180 0 0
## CD74 2.89589 3.35549 0.443406 2.91208 0 0
## CST3 1.48906 2.94985 0.696676 2.25317 0 0plot(dec.pois.pbmc$mean, dec.pois.pbmc$total, pch=16, xlab="Mean of log-expression",
ylab="Variance of log-expression")
curve(metadata(dec.pois.pbmc)$trend(x), col="dodgerblue", add=TRUE)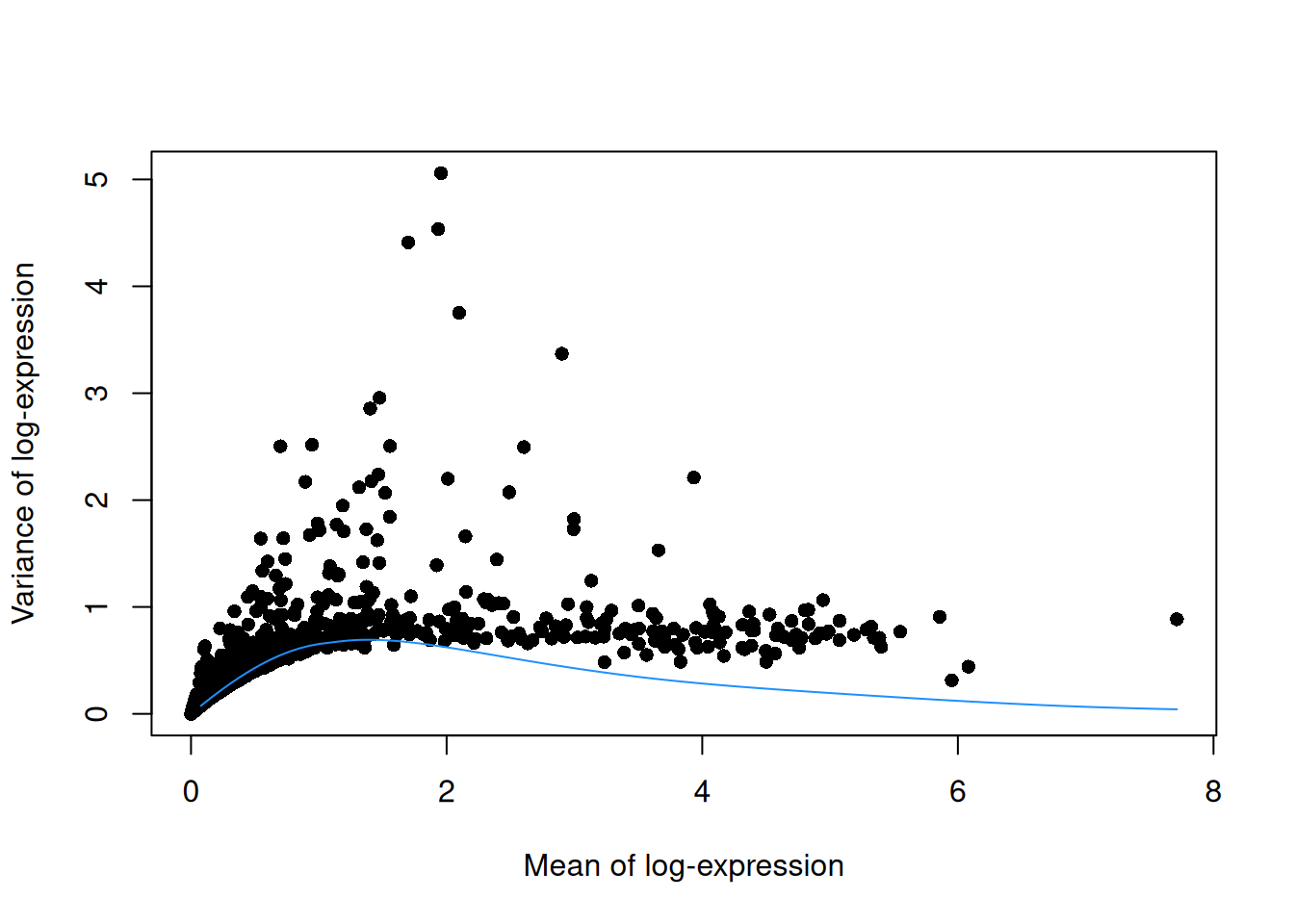
Figure 3.3: Variance of normalized log-expression values for each gene in the PBMC dataset, plotted against the mean log-expression. The blue line represents represents the mean-variance relationship corresponding to Poisson noise.
Interestingly, trends based purely on technical noise tend to yield large biological components for highly-expressed genes. This often includes so-called “house-keeping” genes coding for essential cellular components such as ribosomal proteins, which are considered uninteresting for characterizing cellular heterogeneity. These observations suggest that a more accurate noise model does not necessarily yield a better ranking of HVGs, though one should keep an open mind - house-keeping genes are regularly DE in a variety of conditions (Glare et al. 2002; Nazari, Parham, and Maleki 2015; Guimaraes and Zavolan 2016), and the fact that they have large biological components indicates that there is strong variation across cells that may not be completely irrelevant.
3.4 Handling batch effects
Data containing multiple batches will often exhibit batch effects - see Multi-sample Chapter 1 for more details.
We are usually not interested in HVGs that are driven by batch effects; instead, we want to focus on genes that are highly variable within each batch.
This is naturally achieved by performing trend fitting and variance decomposition separately for each batch.
We demonstrate this approach by treating each plate (block) in the 416B dataset as a different batch, using the modelGeneVarWithSpikes() function.
(The same argument is available in all other variance-modelling functions.)
dec.block.416b <- modelGeneVarWithSpikes(sce.416b, "ERCC", block=sce.416b$block)
head(dec.block.416b[order(dec.block.416b$bio, decreasing=TRUE),1:6])## DataFrame with 6 rows and 6 columns
## mean total tech bio p.value FDR
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## Lyz2 6.61235 13.8619 1.58417 12.2777 0.00000e+00 0.00000e+00
## Ccl9 6.67841 13.2599 1.44553 11.8143 0.00000e+00 0.00000e+00
## Top2a 5.81275 14.0192 2.74575 11.2734 3.96145e-137 8.57006e-135
## Cd200r3 4.83305 15.5909 4.31900 11.2719 1.18818e-54 7.06875e-53
## Ccnb2 5.97999 13.0256 2.46649 10.5591 1.22233e-151 3.02999e-149
## Hbb-bt 4.91683 14.6539 4.12163 10.5322 2.54730e-49 1.35308e-47The use of a batch-specific trend fit is useful as it accommodates differences in the mean-variance trends between batches. This is especially important if batches exhibit systematic technical differences, e.g., differences in coverage or in the amount of spike-in RNA added. In this case, there are only minor differences between the trends in Figure 3.4, which indicates that the experiment was tightly replicated across plates. The analysis of each plate yields estimates of the biological and technical components for each gene, which are averaged across plates to take advantage of information from multiple batches.
par(mfrow=c(1,2))
blocked.stats <- dec.block.416b$per.block
for (i in colnames(blocked.stats)) {
current <- blocked.stats[[i]]
plot(current$mean, current$total, main=i, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(current)
points(curfit$mean, curfit$var, col="red", pch=16)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
}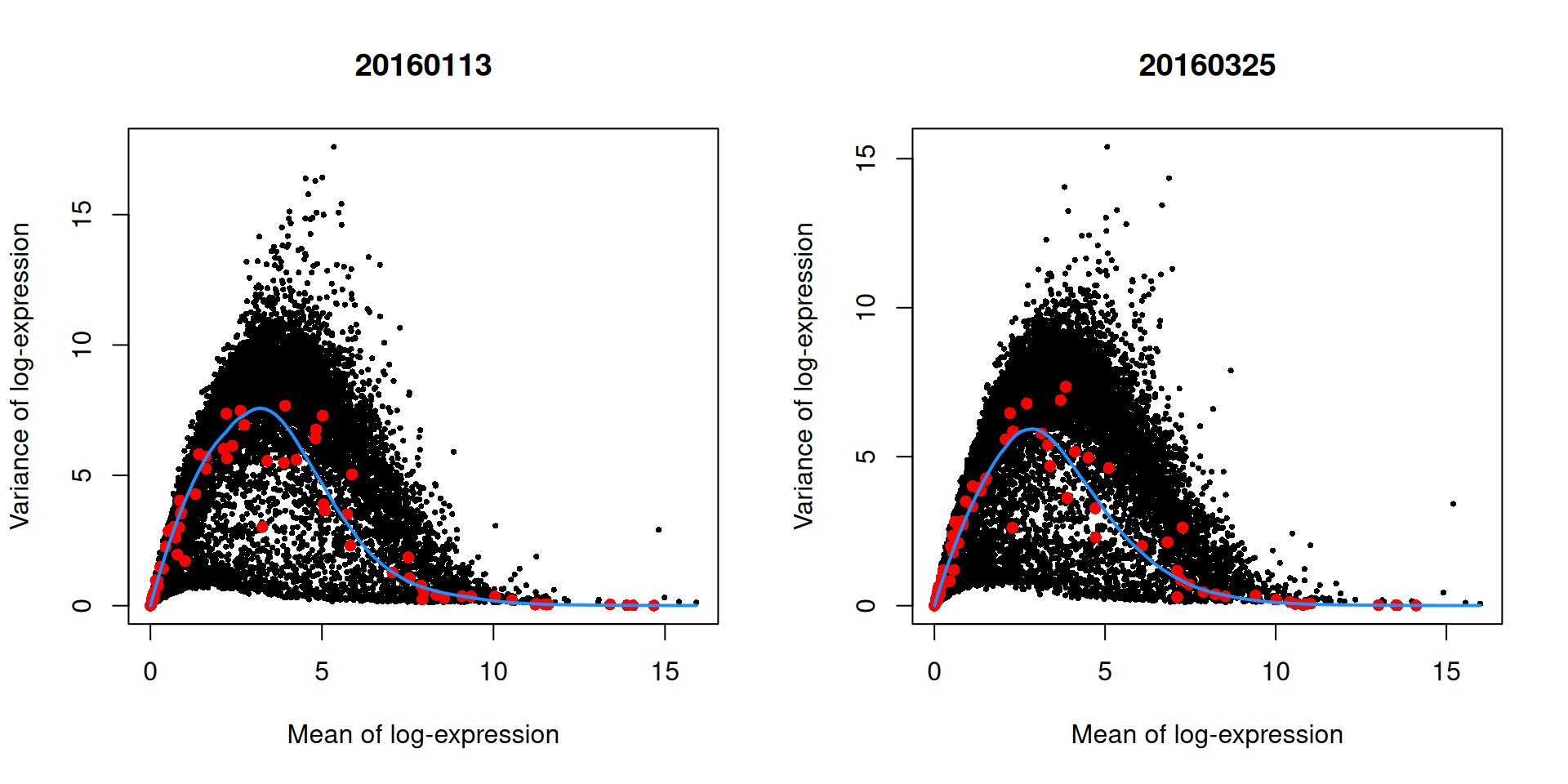
Figure 3.4: Variance in the 416B data set as a function of the mean after blocking on the plate of origin. Each plot represents the results for a single plate, each point represents a gene (black) or spike-in transcript (red) and the blue line represents the trend fitted to all spike-ins.
Alternatively, we might consider using a linear model to account for batch effects and other unwanted factors of variation.
This is more flexible as it can handle multiple factors and continuous covariates, though it is less accurate than block= in the special case of a multi-batch design.
See Advanced Section 3.3 for more details.
As an aside, the wave-like shape observed above is typical of the mean-variance trend for log-expression values. (The same wave is present but much less pronounced for UMI data.) A linear increase in the variance is observed as the mean increases from zero, as larger variances are obviously possible when the counts are not all equal to zero. In contrast, the relative contribution of sampling noise decreases at high abundances, resulting in a downward trend. The peak represents the point at which these two competing effects cancel each other out.
3.5 Selecting highly variable genes
Once we have quantified the per-gene variation, the next step is to select the subset of HVGs to use in downstream analyses. A larger subset will reduce the risk of discarding interesting biological signal by retaining more potentially relevant genes, at the cost of increasing noise from irrelevant genes that might obscure said signal. It is difficult to determine the optimal trade-off for any given application as noise in one context may be useful signal in another. For example, heterogeneity in T cell activation responses is an interesting phenomena (Richard et al. 2018) but may be irrelevant noise in studies that only care about distinguishing the major immunophenotypes.
The most obvious selection strategy is to take the top \(n\) genes with the largest values for the relevant variance metric.
The main advantage of this approach is that the user can directly control the number of genes retained, which ensures that the computational complexity of downstream calculations is easily predicted.
For modelGeneVar() and modelGeneVarWithSpikes(), we would select the genes with the largest biological components.
This is conveniently done for us via getTopHVgs(), as shown below with \(n=1000\).
## chr [1:1000] "LYZ" "S100A9" "S100A8" "HLA-DRA" "CD74" "CST3" "TYROBP" ...The choice of \(n\) also has a fairly straightforward biological interpretation. Recall our trend-fitting assumption that most genes do not exhibit biological heterogeneity; this implies that they are not differentially expressed between cell types or states in our population. If we quantify this assumption into a statement that, e.g., no more than 5% of genes are differentially expressed, we can naturally set \(n\) to 5% of the number of genes. In practice, we usually do not know the proportion of DE genes beforehand so this interpretation just exchanges one unknown for another. Nonetheless, it is still useful as it implies that we should lower \(n\) for less heterogeneous datasets, retaining most of the biological signal without unnecessary noise from irrelevant genes. Conversely, more heterogeneous datasets should use larger values of \(n\) to preserve secondary factors of variation beyond those driving the most obvious HVGs.
The main disadvantage of this approach that it turns HVG selection into a competition between genes, whereby a subset of very highly variable genes can push other informative genes out of the top set. This can be problematic for analyses of highly heterogeneous populations if the loss of important markers prevents the resolution of certain subpopulations. In the most extreme example, consider a situation where a single subpopulation is very different from the others. In such cases, the top set will be dominated by differentially expressed genes involving that distinct subpopulation, compromising resolution of heterogeneity between the other populations. (This can be recovered with a nested analysis, as discussed in Section 5.5, but we would prefer to avoid the problem in the first place.)
Another potential concern with this approach is the fact that the choice of \(n\) is fairly arbitrary, with any value from 500 to 5000 considered “reasonable”. We have chosen \(n=1000\) in the code above though there is no particular a priori reason for doing so. Our recommendation is to simply pick an arbitrary \(n\) and proceed with the rest of the analysis, with the intention of testing other choices later, rather than spending much time worrying about obtaining the “optimal” value. Alternatively, we may pick one of the other selection strategies discussed in Advanced Section 3.5.
3.6 Putting it all together
The code chunk below will select the top 10% of genes with the highest biological components.
## chr [1:1262] "LYZ" "S100A9" "S100A8" "HLA-DRA" "CD74" "CST3" "TYROBP" ...We then have several options to enforce our HVG selection on the rest of the analysis.
We can subset the
SingleCellExperimentto only retain our selection of HVGs. This ensures that downstream methods will only use these genes for their calculations. The downside is that the non-HVGs are discarded from the newSingleCellExperiment, making it slightly more inconvenient to interrogate the full dataset for interesting genes that are not HVGs.## [1] 1262 4080We can keep the original
SingleCellExperimentobject and specify the genes to use for downstream functions via an extra argument likesubset.row=. This is useful if the analysis uses multiple sets of HVGs at different steps, whereby one set of HVGs can be easily swapped for another in specific steps.# Performing PCA only on the chosen HVGs. library(scater) sce.pbmc <- runPCA(sce.pbmc, subset_row=chosen) reducedDimNames(sce.pbmc)## [1] "PCA"This approach is facilitated by the
rowSubset()utility, which allows us to easily store one or more sets of interest in ourSingleCellExperiment. By doing so, we avoid the need to keep track of a separatechosenvariable and ensure that our HVG set is synchronized with any downstream row subsetting ofsce.pbmc.rowSubset(sce.pbmc) <- chosen # stored in the default 'subset'. rowSubset(sce.pbmc, "HVGs.more") <- getTopHVGs(dec.pbmc, prop=0.2) rowSubset(sce.pbmc, "HVGs.less") <- getTopHVGs(dec.pbmc, prop=0.3) colnames(rowData(sce.pbmc))## [1] "ID" "Symbol" "subset" "HVGs.more" "HVGs.less"It can be inconvenient to repeatedly specify the desired feature set across steps, so some downstream functions will automatically subset to the default
rowSubset()if present in theSingleCellExperiment. However, we find that it is generally safest to be explicit about which set is being used for a particular step.We can have our cake and eat it too by (ab)using the “alternative Experiment” system in the
SingleCellExperimentclass. Initially designed for storing alternative features like spike-ins or antibody tags, we can instead use it to hold our full dataset while we perform our downstream operations conveniently on the HVG subset. This avoids book-keeping problems in long analyses when the original dataset is not synchronized with the HVG subsetted data.## [1] "original"
Session Info
R version 4.5.1 Patched (2025-08-23 r88802)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 24.04.3 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.22-bioc/R/lib/libRblas.so
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.12.0 LAPACK version 3.12.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: America/New_York
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] scater_1.38.0 ggplot2_4.0.0
[3] scran_1.38.0 scuttle_1.20.0
[5] SingleCellExperiment_1.32.0 SummarizedExperiment_1.40.0
[7] Biobase_2.70.0 GenomicRanges_1.62.0
[9] Seqinfo_1.0.0 IRanges_2.44.0
[11] S4Vectors_0.48.0 BiocGenerics_0.56.0
[13] generics_0.1.4 MatrixGenerics_1.22.0
[15] matrixStats_1.5.0 BiocStyle_2.38.0
[17] rebook_1.20.0
loaded via a namespace (and not attached):
[1] tidyselect_1.2.1 viridisLite_0.4.2 vipor_0.4.7
[4] dplyr_1.1.4 farver_2.1.2 viridis_0.6.5
[7] filelock_1.0.3 S7_0.2.0 fastmap_1.2.0
[10] bluster_1.20.0 XML_3.99-0.19 digest_0.6.37
[13] rsvd_1.0.5 lifecycle_1.0.4 cluster_2.1.8.1
[16] statmod_1.5.1 magrittr_2.0.4 compiler_4.5.1
[19] rlang_1.1.6 sass_0.4.10 tools_4.5.1
[22] igraph_2.2.1 yaml_2.3.10 knitr_1.50
[25] S4Arrays_1.10.0 dqrng_0.4.1 DelayedArray_0.36.0
[28] RColorBrewer_1.1-3 abind_1.4-8 BiocParallel_1.44.0
[31] withr_3.0.2 CodeDepends_0.6.6 grid_4.5.1
[34] beachmat_2.26.0 edgeR_4.8.0 scales_1.4.0
[37] dichromat_2.0-0.1 cli_3.6.5 rmarkdown_2.30
[40] metapod_1.18.0 ggbeeswarm_0.7.2 cachem_1.1.0
[43] parallel_4.5.1 BiocManager_1.30.26 XVector_0.50.0
[46] vctrs_0.6.5 Matrix_1.7-4 jsonlite_2.0.0
[49] dir.expiry_1.18.0 bookdown_0.45 BiocSingular_1.26.0
[52] BiocNeighbors_2.4.0 ggrepel_0.9.6 beeswarm_0.4.0
[55] irlba_2.3.5.1 locfit_1.5-9.12 limma_3.66.0
[58] jquerylib_0.1.4 glue_1.8.0 codetools_0.2-20
[61] gtable_0.3.6 ScaledMatrix_1.18.0 tibble_3.3.0
[64] pillar_1.11.1 rappdirs_0.3.3 htmltools_0.5.8.1
[67] graph_1.88.0 R6_2.6.1 evaluate_1.0.5
[70] lattice_0.22-7 bslib_0.9.0 Rcpp_1.1.0
[73] gridExtra_2.3 SparseArray_1.10.1 xfun_0.54
[76] pkgconfig_2.0.3 References
Glare, E. M., M. Divjak, M. J. Bailey, and E. H. Walters. 2002. “beta-Actin and GAPDH housekeeping gene expression in asthmatic airways is variable and not suitable for normalising mRNA levels.” Thorax 57 (9): 765–70.
Guimaraes, J. C., and M. Zavolan. 2016. “Patterns of ribosomal protein expression specify normal and malignant human cells.” Genome Biol. 17 (1): 236.
Lun, A. T. L., D. J. McCarthy, and J. C. Marioni. 2016. “A Step-by-Step Workflow for Low-Level Analysis of Single-Cell RNA-seq Data.” F1000Res. 5 (August).
Nazari, F., A. Parham, and A. F. Maleki. 2015. “GAPDH, -actin and -microglobulin, as three common reference genes, are not reliable for gene expression studies in equine adipose- and marrow-derived mesenchymal stem cells.” J Anim Sci Technol 57: 18.
Richard, A. C., A. T. L. Lun, W. W. Y. Lau, B. Gottgens, J. C. Marioni, and G. M. Griffiths. 2018. “T cell cytolytic capacity is independent of initial stimulation strength.” Nat. Immunol. 19 (8): 849–58.