# HCA human bone marrow (10X Genomics)
## Introduction
Here, we use an example dataset from the [Human Cell Atlas immune cell profiling project on bone marrow](https://preview.data.humancellatlas.org), which contains scRNA-seq data for 380,000 cells generated using the 10X Genomics technology.
This is a fairly big dataset that represents a good use case for the techniques in [Advanced Chapter 14](http://bioconductor.org/books/3.21/OSCA.advanced/dealing-with-big-data.html#dealing-with-big-data).
## Data loading
This dataset is loaded via the *[HCAData](https://bioconductor.org/packages/3.21/HCAData)* package, which provides a ready-to-use `SingleCellExperiment` object.
``` r
library(HCAData)
sce.bone <- HCAData('ica_bone_marrow', as.sparse=TRUE)
sce.bone$Donor <- sub("_.*", "", sce.bone$Barcode)
```
We use symbols in place of IDs for easier interpretation later.
``` r
library(EnsDb.Hsapiens.v86)
rowData(sce.bone)$Chr <- mapIds(EnsDb.Hsapiens.v86, keys=rownames(sce.bone),
column="SEQNAME", keytype="GENEID")
library(scater)
rownames(sce.bone) <- uniquifyFeatureNames(rowData(sce.bone)$ID,
names = rowData(sce.bone)$Symbol)
```
## Quality control
Cell calling was not performed (see [here](https://s3.amazonaws.com/preview-ica-expression-data/Brief+ICA+Read+Me.pdf)) so we will perform QC using all metrics and block on the donor of origin during outlier detection.
We perform the calculation across multiple cores to speed things up.
``` r
library(BiocParallel)
bpp <- MulticoreParam(8)
sce.bone <- unfiltered <- addPerCellQC(sce.bone, BPPARAM=bpp,
subsets=list(Mito=which(rowData(sce.bone)$Chr=="MT")))
qc <- quickPerCellQC(colData(sce.bone), batch=sce.bone$Donor,
sub.fields="subsets_Mito_percent")
sce.bone <- sce.bone[,!qc$discard]
```
``` r
unfiltered$discard <- qc$discard
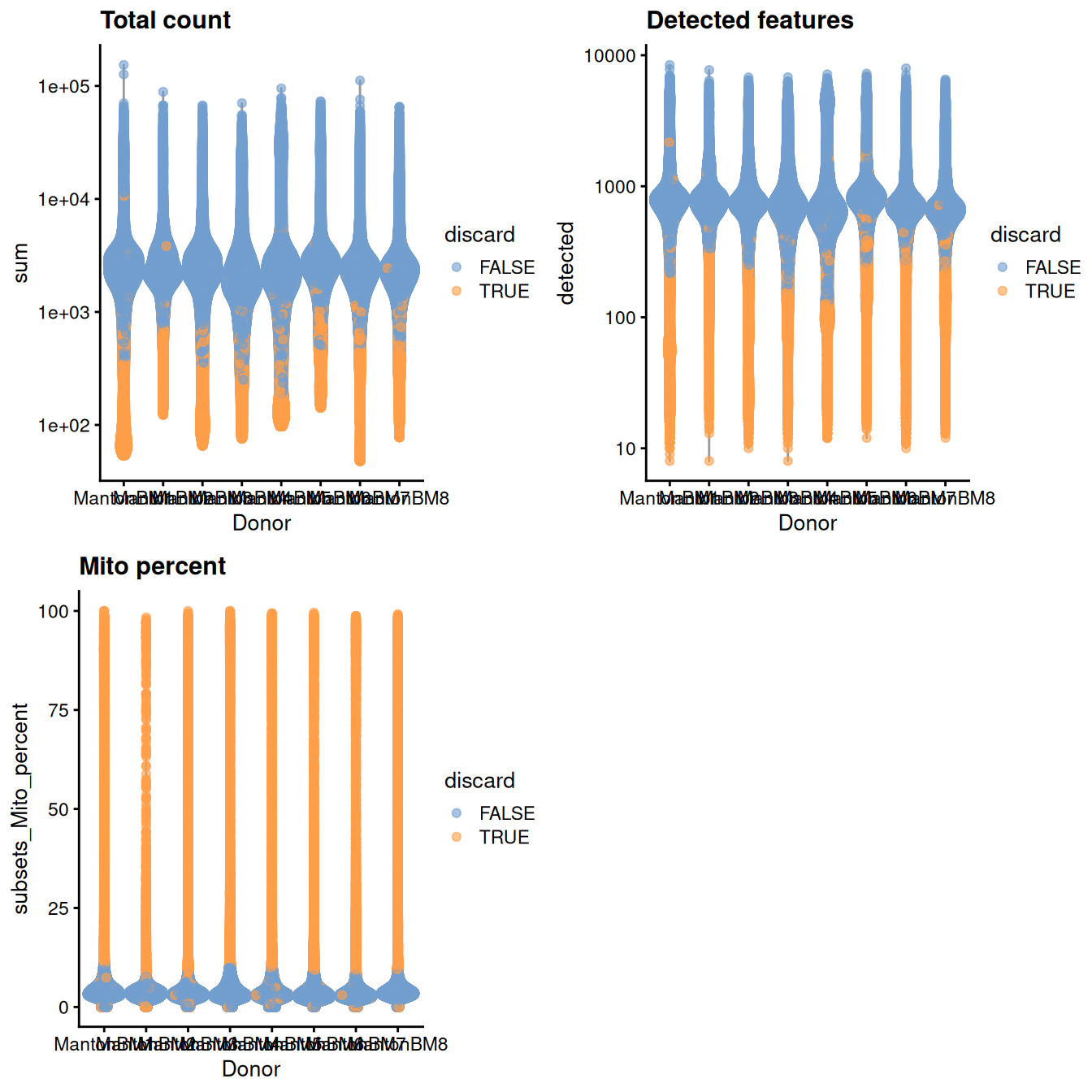
gridExtra::grid.arrange(
plotColData(unfiltered, x="Donor", y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, x="Donor", y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, x="Donor", y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
ncol=2
)
```
(\#fig:unref-hca-bone-qc)Distribution of QC metrics in the HCA bone marrow dataset. Each point represents a cell and is colored according to whether it was discarded.
``` r
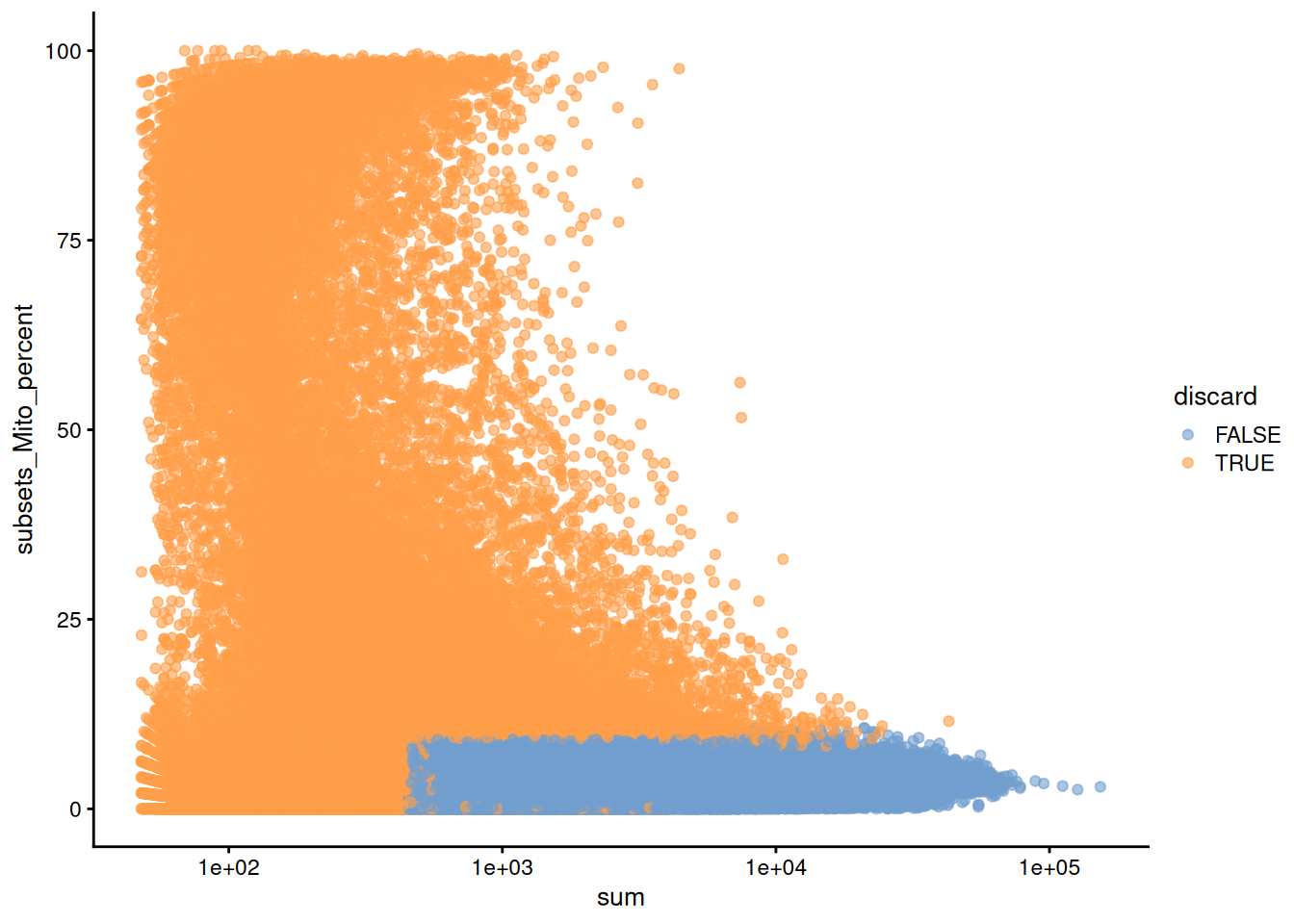
plotColData(unfiltered, x="sum", y="subsets_Mito_percent",
colour_by="discard") + scale_x_log10()
```
(\#fig:unref-hca-bone-mito)Percentage of mitochondrial reads in each cell in the HCA bone marrow dataset compared to its total count. Each point represents a cell and is colored according to whether that cell was discarded.
## Normalization
For a minor speed-up, we use already-computed library sizes rather than re-computing them from the column sums.
``` r
sce.bone <- logNormCounts(sce.bone, size_factors = sce.bone$sum)
```
``` r
summary(sizeFactors(sce.bone))
```
```
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.05 0.47 0.65 1.00 0.89 42.38
```
## Variance modeling
We block on the donor of origin to mitigate batch effects during HVG selection.
We select a larger number of HVGs to capture any batch-specific variation that might be present.
``` r
library(scran)
set.seed(1010010101)
dec.bone <- modelGeneVarByPoisson(sce.bone,
block=sce.bone$Donor, BPPARAM=bpp)
top.bone <- getTopHVGs(dec.bone, n=5000)
```
``` r
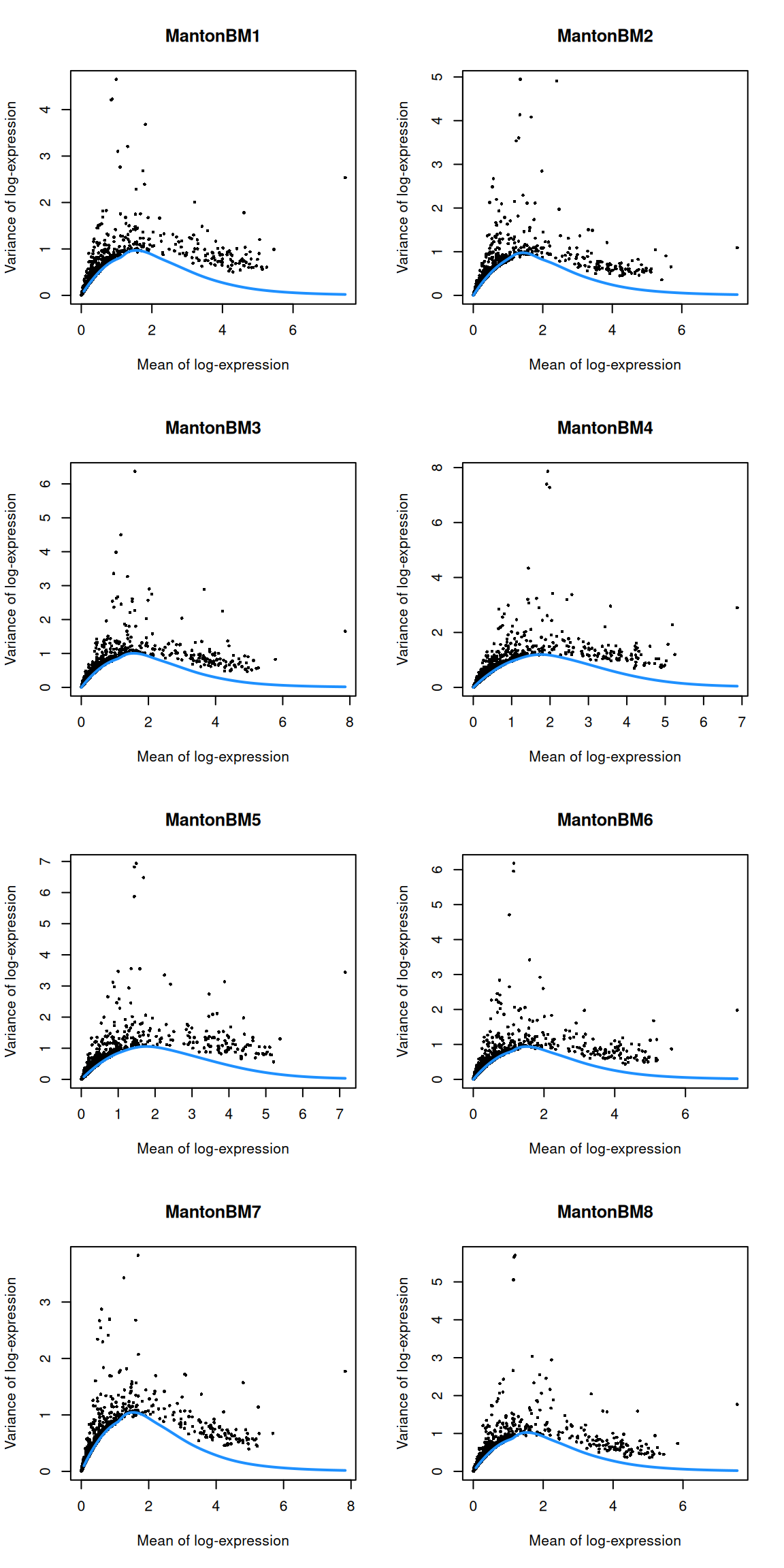
par(mfrow=c(4,2))
blocked.stats <- dec.bone$per.block
for (i in colnames(blocked.stats)) {
current <- blocked.stats[[i]]
plot(current$mean, current$total, main=i, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(current)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
}
```
(\#fig:unref-hca-bone-var)Per-gene variance as a function of the mean for the log-expression values in the HCA bone marrow dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to the variances.
## Data integration
Here we use multiple cores, randomized SVD and approximate nearest-neighbor detection to speed up this step.
``` r
library(batchelor)
library(BiocNeighbors)
set.seed(1010001)
merged.bone <- fastMNN(sce.bone, batch = sce.bone$Donor, subset.row = top.bone,
BSPARAM=BiocSingular::RandomParam(deferred = TRUE),
BNPARAM=AnnoyParam(),
BPPARAM=bpp)
reducedDim(sce.bone, 'MNN') <- reducedDim(merged.bone, 'corrected')
```
We use the percentage of variance lost as a diagnostic measure:
``` r
metadata(merged.bone)$merge.info$lost.var
```
```
## MantonBM1 MantonBM2 MantonBM3 MantonBM4 MantonBM5 MantonBM6 MantonBM7
## [1,] 0.007133 0.006508 0.000000 0.000000 0.000000 0.000000 0.000000
## [2,] 0.006314 0.006883 0.023528 0.000000 0.000000 0.000000 0.000000
## [3,] 0.005117 0.003096 0.005115 0.019703 0.000000 0.000000 0.000000
## [4,] 0.001991 0.001888 0.001890 0.001766 0.023451 0.000000 0.000000
## [5,] 0.002391 0.001914 0.001735 0.002805 0.002563 0.023692 0.000000
## [6,] 0.003053 0.003180 0.002958 0.002522 0.003211 0.003342 0.024807
## [7,] 0.001826 0.001591 0.002290 0.001881 0.001473 0.002174 0.001908
## MantonBM8
## [1,] 0.00000
## [2,] 0.00000
## [3,] 0.00000
## [4,] 0.00000
## [5,] 0.00000
## [6,] 0.00000
## [7,] 0.03235
```
## Dimensionality reduction
We set `external_neighbors=TRUE` to replace the internal nearest neighbor search in the UMAP implementation with our parallelized approximate search.
We also set the number of threads to be used in the UMAP iterations.
``` r
set.seed(01010100)
sce.bone <- runUMAP(sce.bone, dimred="MNN",
external_neighbors=TRUE,
BNPARAM=AnnoyParam(),
BPPARAM=bpp,
n_threads=bpnworkers(bpp))
```
## Clustering
Graph-based clustering generates an excessively large intermediate graph so we will instead use a two-step approach with $k$-means.
We generate 1000 small clusters that are subsequently aggregated into more interpretable groups with a graph-based method.
If more resolution is required, we can increase `centers` in addition to using a lower `k` during graph construction.
``` r
library(bluster)
set.seed(1000)
colLabels(sce.bone) <- clusterRows(reducedDim(sce.bone, "MNN"),
TwoStepParam(KmeansParam(centers=1000), NNGraphParam(k=5)))
table(colLabels(sce.bone))
```
```
##
## 1 2 3 4 5 6 7 8 9 10 11 12 13
## 18859 15812 36360 47699 26528 10869 65650 18584 35321 8009 14930 3601 4206
## 14 15 16
## 3155 4824 2318
```
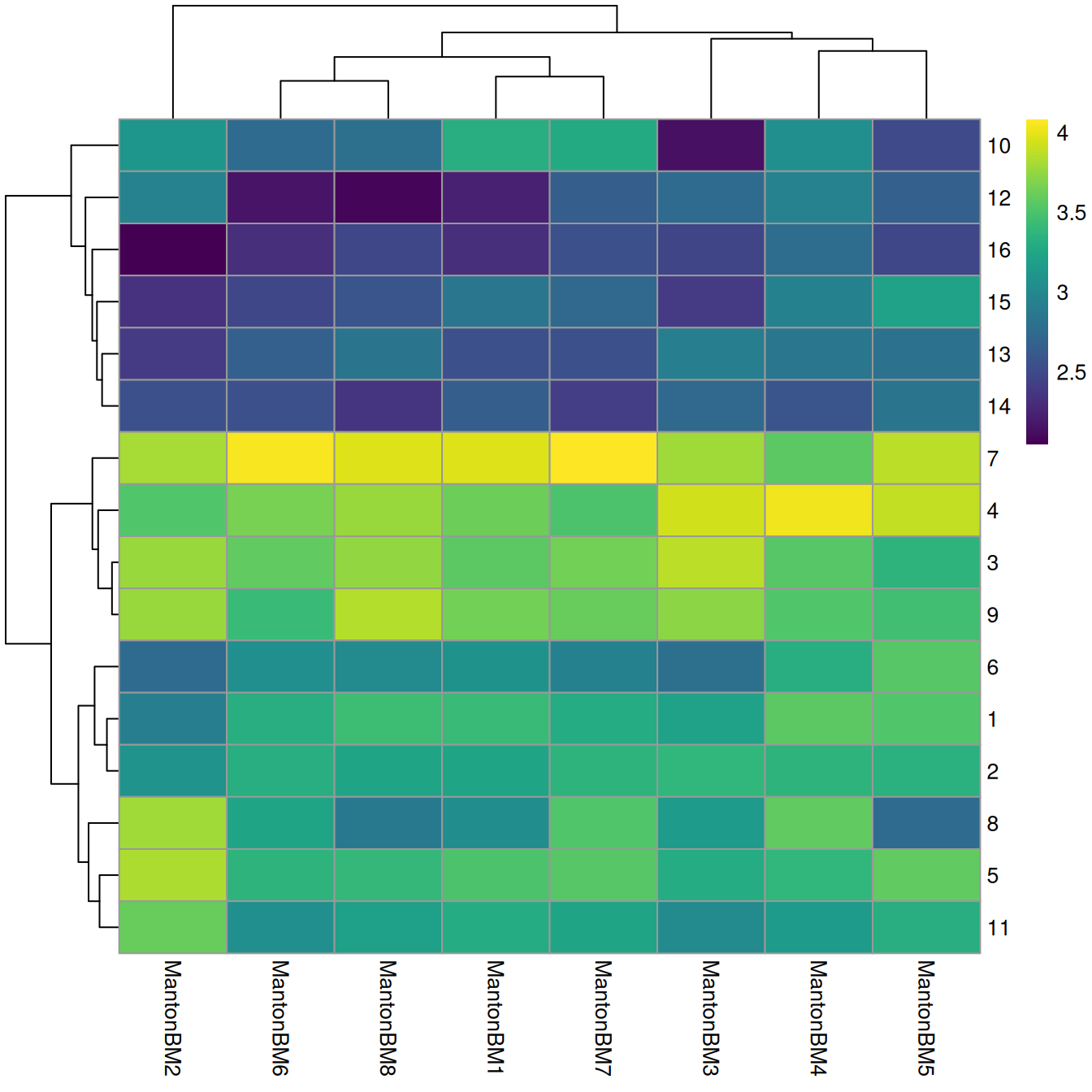
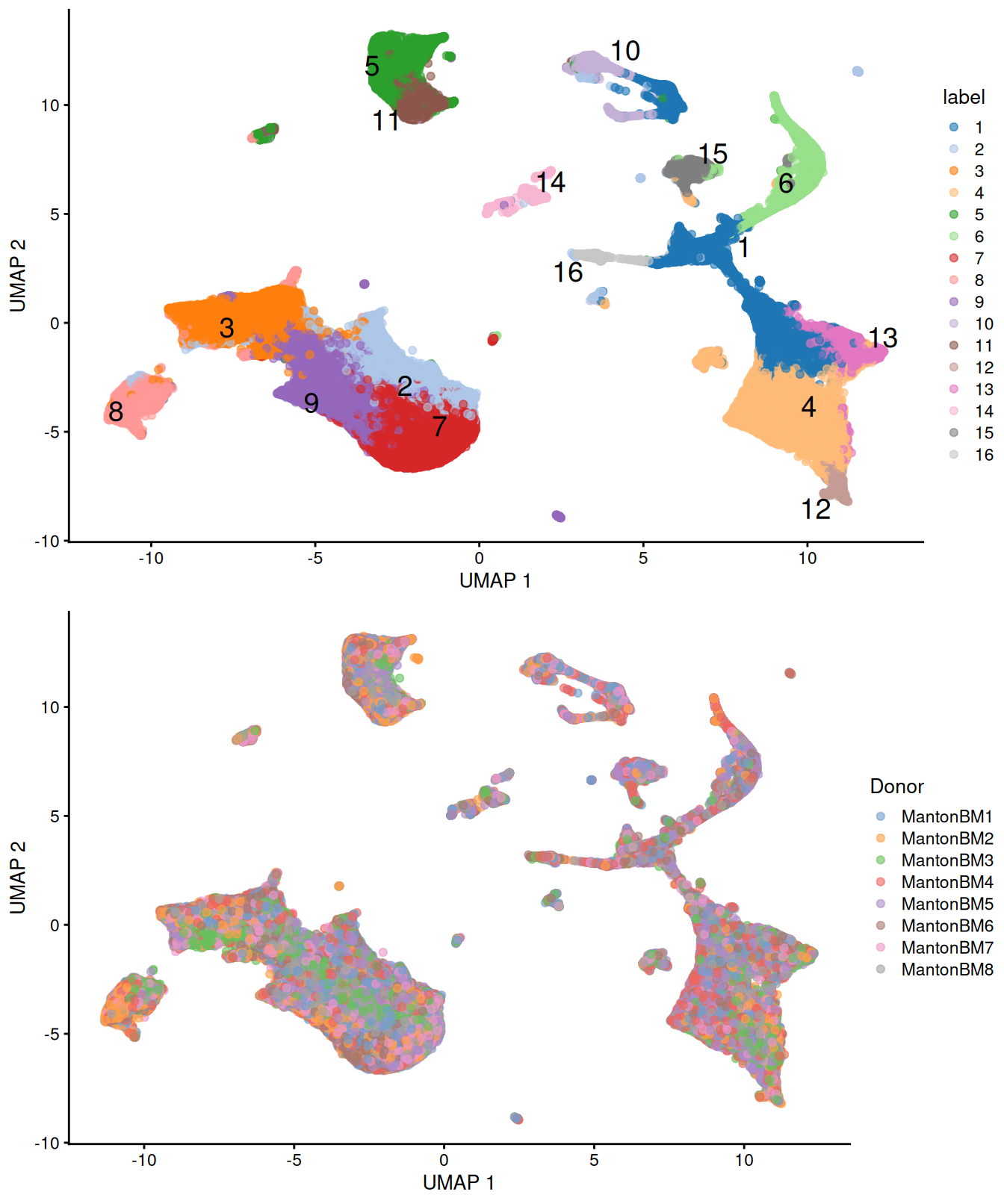
We observe mostly balanced contributions from different samples to each cluster (Figure \@ref(fig:unref-hca-bone-ab)), consistent with the expectation that all samples are replicates from different donors.
``` r
tab <- table(Cluster=colLabels(sce.bone), Donor=sce.bone$Donor)
library(pheatmap)
pheatmap(log10(tab+10), color=viridis::viridis(100))
```
(\#fig:unref-hca-bone-ab)Heatmap of log~10~-number of cells in each cluster (row) from each sample (column).
(\#fig:unref-hca-bone-umap)UMAP plots of the HCA bone marrow dataset after merging. Each point represents a cell and is colored according to the assigned cluster (top) or the donor of origin (bottom).
## Differential expression
We identify marker genes for each cluster while blocking on the donor.
``` r
markers.bone <- findMarkers(sce.bone, block = sce.bone$Donor,
direction = 'up', lfc = 1, BPPARAM=bpp)
```
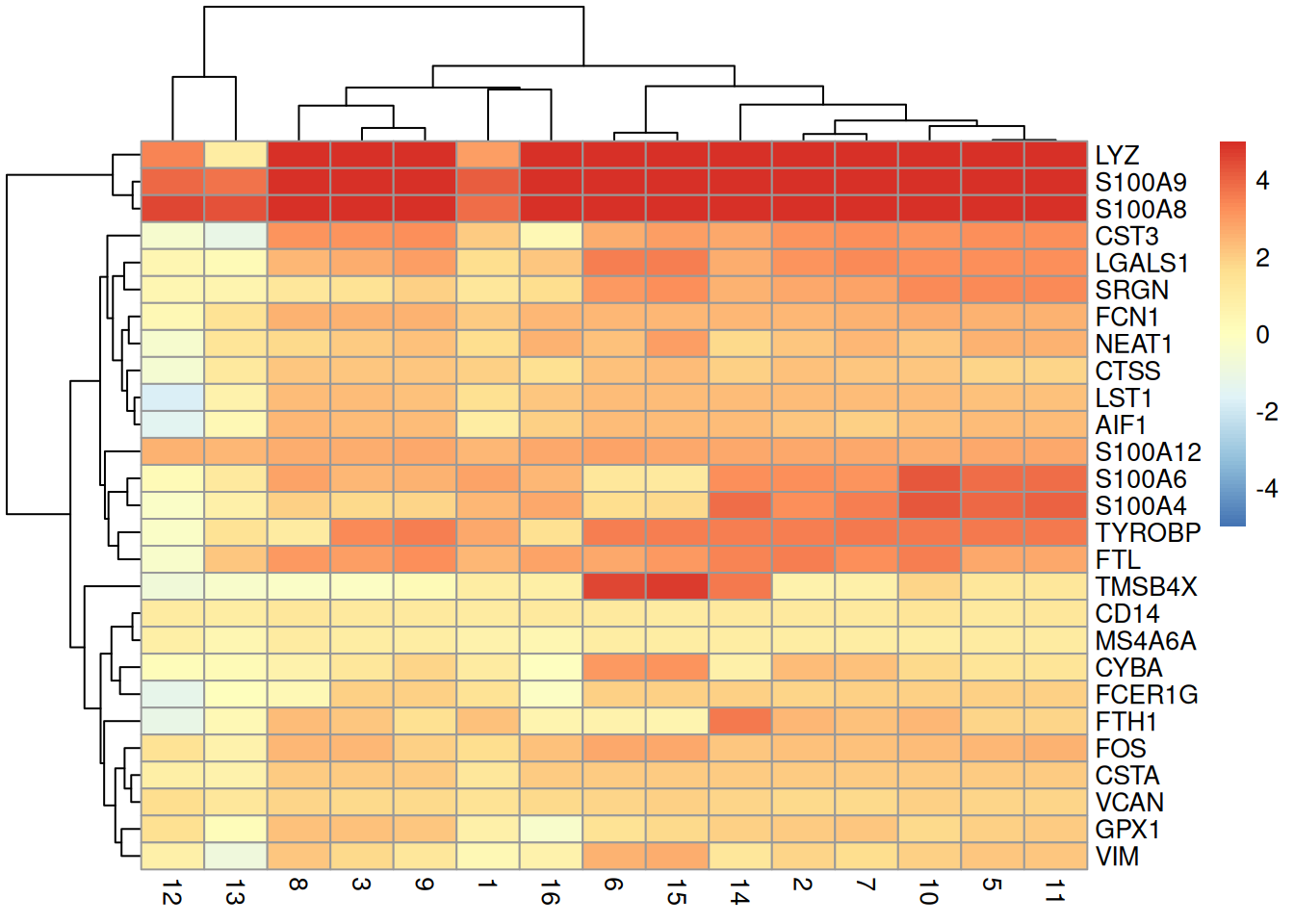
We visualize the top markers for a randomly chosen cluster using a "dot plot" in Figure \@ref(fig:unref-hca-bone-dotplot).
The presence of upregulated genes like _LYZ_, _S100A8_ and _VCAN_ is consistent with a monocyte identity for this cluster.
``` r
top.markers <- markers.bone[["4"]]
best <- top.markers[top.markers$Top <= 10,]
lfcs <- getMarkerEffects(best)
library(pheatmap)
pheatmap(lfcs, breaks=seq(-5, 5, length.out=101))
```
(\#fig:unref-hca-bone-dotplot)Heatmap of log~2~-fold changes for the top marker genes (rows) of cluster 4 compared to all other clusters (columns).
## Cell type classification
We perform automated cell type classification using a reference dataset to annotate each cluster based on its pseudo-bulk profile.
This is faster than the per-cell approaches described in Chapter \@ref(cell-type-annotation) at the cost of the resolution required to detect heterogeneity inside a cluster.
Nonetheless, it is often sufficient for a quick assignment of cluster identity, and indeed, cluster 4 is also identified as consisting of monocytes from this analysis.
``` r
se.aggregated <- sumCountsAcrossCells(sce.bone, id=colLabels(sce.bone), BPPARAM=bpp)
library(celldex)
hpc <- HumanPrimaryCellAtlasData()
library(SingleR)
anno.single <- SingleR(se.aggregated, ref = hpc, labels = hpc$label.main,
assay.type.test="sum")
anno.single
```
```
## DataFrame with 16 rows and 4 columns
## scores labels delta.next pruned.labels
##
## 1 0.384401:0.751148:0.651234:... GMP 0.0913786 GMP
## 2 0.343557:0.567261:0.479100:... T_cells 0.4298632 T_cells
## 3 0.323043:0.647364:0.558334:... T_cells 0.0959201 T_cells
## 4 0.299294:0.745584:0.535751:... Monocyte 0.2935059 Monocyte
## 5 0.310761:0.672644:0.540285:... B_cell 0.6024293 B_cell
## ... ... ... ... ...
## 12 0.294203:0.707235:0.528198:... Monocyte 0.3586359 Monocyte
## 13 0.343741:0.731258:0.600058:... Monocyte 0.1019188 NA
## 14 0.369798:0.652467:0.582201:... B_cell 0.1976631 NA
## 15 0.378580:0.690882:0.781190:... MEP 0.0614135 MEP
## 16 0.333963:0.679341:0.559147:... GMP 0.1114087 GMP
```
## Session Info {-}